- 热门化合物 :
- 物质11
- 2-丙炔-1-醇
- americium,trifluoride
- neptunium hexafluoride
- neptunium tetrachloride
nature!又一篇光催化成果登刊!
图2:反应设计及机理分析。a,通过光催化三苯基膦介导的水活化析氢。b, PR3-OH中间体转化成闭壳非活化烯烃的一般机理。c、通过密度泛函理论(DFT)计算得到的各种PR3-OH中间体中1a转移...
水的化学活化是能源研究中的热点,它能够将地球上丰富的资源转化为具有增值潜力的化合物。而氢被视为未来的能源之一,特别是在低碳环保的环境下以对气候友好的方式生成。此外,氢也是生成活性成分和其他基础物质所必需的。要产生氢气,可以通过一系列化学过程将水(H2O)转化为氢气(H2)。然而,由于水分子的稳定性高,将其分解成氧和氢目前面临巨大的挑战。
鉴于此,近期,由明斯特大学有机化学研究所Armido Studer教授带领的研究小组在Nature期刊上发表了题为“Photocatalytic phosphine-mediated water activation for radical hydrogenation”的研究成果。
概述
研究展示了一种在温和条件下利用光催化膦介导的自由基过程对水进行活化的方法。该过程产生了一个不含金属的PR3-H2O自由基阳离子中间体,在其中两个氢原子经过连续的异裂(H+)和均裂(H•)将其转化为其他化合物。PR3-OH自由基中间体提供了一个理想的平台,模拟了自由氢原子的反应性,并可直接转移到闭壳π体系,如活化烯烃、未活化烯烃、萘和喹啉衍生物等。生成的H加合物C自由基最终通过巯基辅助催化剂还原,从而实现整体的π体系转移氢化反应,最终产物中包含来自水的两个氢原子。这一反应的热力学驱动力是在磷膦氧副产物中形成的强P=O键。通过实验研究和密度泛函理论计算支持了PR3-OH中间体的氢原子转移作为自由基氢化过程中的关键步骤。
图文导读
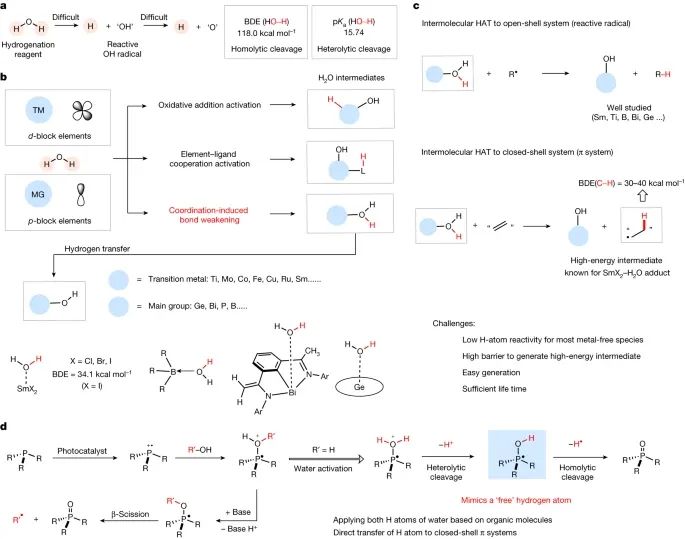
水可以通过一系列化学过程转化为有价值的氢气(H2),这使得它可以作为一种潜在的转移加氢试剂用于还原不饱和化合物。然而,水活化的主要挑战来自两个特征(图1a)。水的去质子化(pKa = 15.74)在合适的碱基下很容易实现,但由于O-H键的高键解离能(BDE = 118 kcal mol -1 ), H2O具有较高的热力学稳定性,因此均解裂解具有挑战性。此外,OH自由基(羟基自由基),如果产生,不能提供第二个氢原子,因为这将导致一个高能氧原子。因此,需要催化剂或介质,目前的水活化方法遵循三种不同的策略:氧化加成活化、金属配体协同活化和配位诱导的键减弱(图1b)。前两种方法主要集中在d块过渡金属的使用上,而处理主族元素的转换仍然非常罕见。通过生成水加合物并利用配位诱导键减弱的概念,过渡金属配合物和具有空d或p轨道的主族元素已经展示了一些例子。这些H2O加合物的形成增加了酸度,并且大多数作为质子供体参与反应。已知的例子包括钐(II)、钛(III)、硼和铋(II)等化合物,它们可以实现O-H键的均裂性。这些活性水配合物能够减少活性自由基的数量,生成闭壳产物,在热力学上促使氢原子转移(HAT)反应发生(如图1c所示)。
图1:背景与概念。a、水作为潜在的转移氢化试剂;BDE和pKa。b,不同水活化模式概述及活化H2O加合物举例。c,通过水加合物的分子间HAT还原自由基和HAT还原成闭壳π体系。
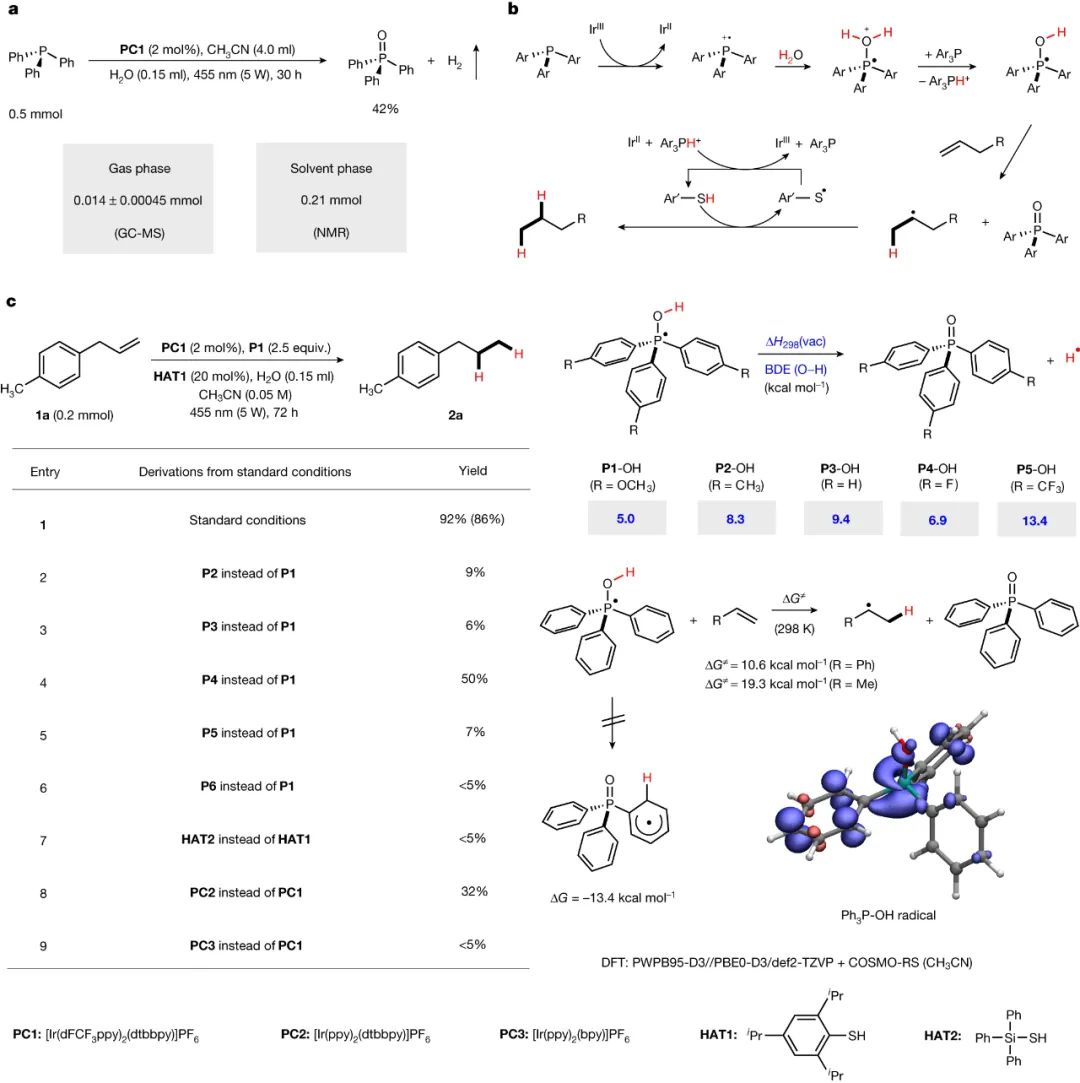
考虑到光氧化还原催化中磷化氢自由基阳离子的可调介质特性,可以通过脱氧生成碳中心和杂原子中心的自由基(见图1d)。因此,研究设计了一种相关方法,利用磷化氢自由基阳离子的单占据分子轨道与水相互作用,通过光氧化还原催化实现水活化。为了验证PR3-OH中间体的反应设计和H原子反应性,首先研究了三苯基膦(PPh3)介导的析氢反应。实验在5 W蓝色发光二极管(LED)照射下,使用Ir(dF(CF3)ppy)2(dtbbpy)(其中dF(CF3)ppy为2-(2,4-二氟苯基)-5-三氟甲基吡啶,dbbpy为4,4′-二叔丁基-2,2′-联吡啶,PC1)作为光催化剂,在乙腈中以H2O为氢源,成功验证了溶液和气相中H2的产生,并同时生成副产物三苯基氧化膦(见图2a)。同时还进一步研究了未活化的烯烃与水作为氢供体进行的转移氢化反应(见图2b)。
图2:反应设计及机理分析。a,通过光催化三苯基膦介导的水活化析氢。b, PR3-OH中间体转化成闭壳非活化烯烃的一般机理。c、通过密度泛函理论(DFT)计算得到的各种PR3-OH中间体中1a转移加氢和O-H键BDE的反应优化。
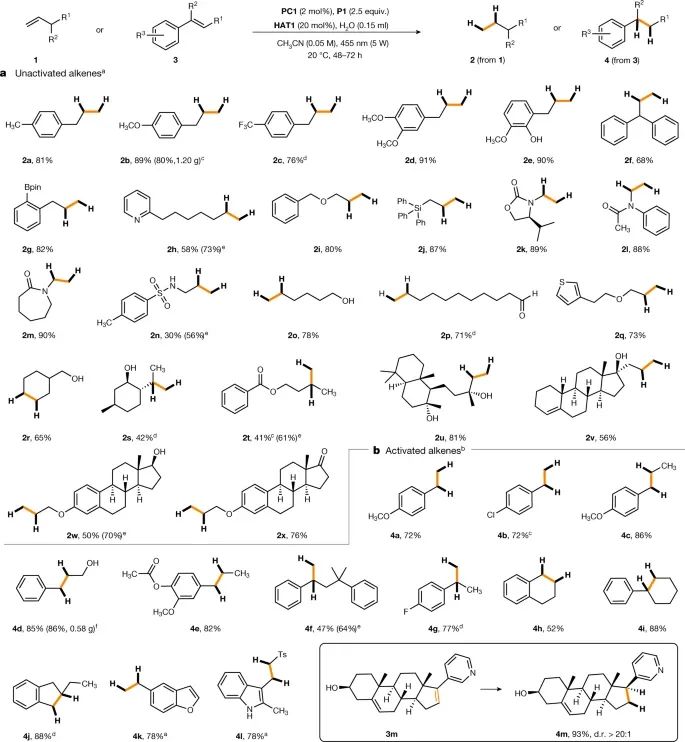
随着优化条件的确定,可以高效地对带有不同官能团的未激活烯烃进行氢化。苄基芳烃在芳香环上携带电子供体或电子受体取代基时,可以以高产率得到相应的丙基芳烃(图3a)。自由基氢化可以与常用的官能团相容,包括硼酸磷酰可醇酯(2g)、吡啶(2h)、醚(2i)、三苯基硅(2j)、噁唑烷酮(2k)、酰胺(2l和2m)、磺酰胺(2n)、自由醇(2o和2r)、醛(2p)和噻吩(2q)基团。虽然双取代烯烃的还原产率较中等(2s和2t),但也可以被还原。通过温和的方法,研究对具有末端未激活双键的复杂化合物进行后期氢化,实验通过对沉香醇(2u)、丙烯诺醇(2v)、β-雌二醇(2w)和酮类衍生物(2x)进行还原,得以证明。值得注意的是,丙烯诺醇1v的还原产物2v具有完全的化学选择性,环中的三取代烯烃基团保持未反应状态。更具反应活性的苯乙烯衍生物可利用PPh3(P3)作为H原子传递剂进行便捷氢化,并且反应时间可以缩短(48小时)。各种苯乙烯衍生物,包括α-和β-取代同系物以及三取代系统,可以以中等至优异的产率还原(图3b)。类固醇化合物3m被完全的化学选择性和立体选择性地氢化。
图3:转移氢化底物范围。a、未活化烯烃的还原。b,苯乙烯衍生物的还原。
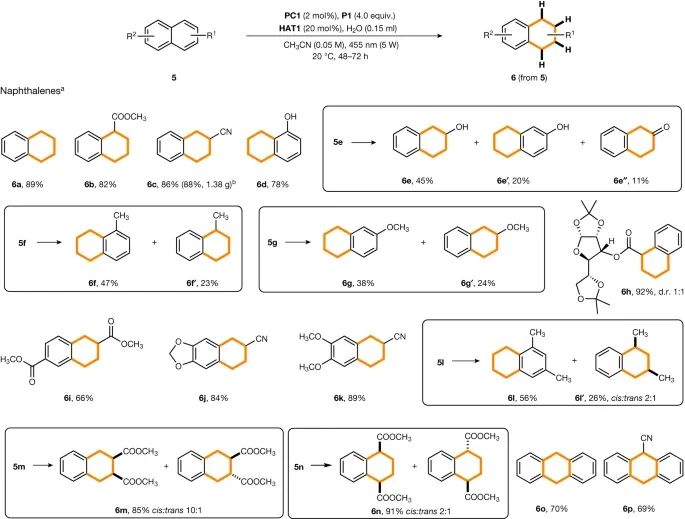
研究对萘及其衍生物进行的氢化反应(图4)。实验结果表明,酯、腈和羟基取代的萘可以高产率地氢化成相应的四氢萘丙烷。在这些情况下,没有观察到第二个芳香环的氢化反应。此外,带有电子吸引基团的萘在取代基团上发生去芳香化。
图4:萘的还原。
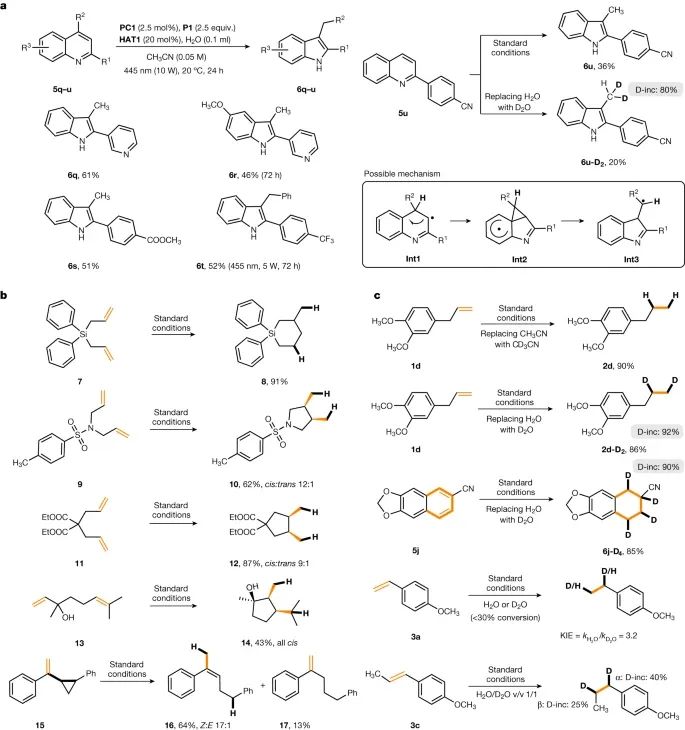
研究展示了对喹啉的骨架重排反应(图5)。通过这种光催化磷胺介导的水活化体系,在产生自由基中间体的过程中实现了喹啉的直接骨架编辑。该反应不能通过过渡金属介导的氢化过程实现,并且是其他自由基还原体系所不具备的。实验结果显示,2-取代喹啉经过骨架重排反应后可以得到相应的2,3-二取代吲哚产物。同时,2,4-二取代的喹啉也能发生类似的转化生成吲哚产物。
图5:应用和机理研究。
小结
综上所述,在室温下,原位生成的PR3-OH自由基可以作为HAT试剂与各种烯烃和萘反应,生成相应的H原子加成加合自由基。关键的PR3-OH自由基,可以被认为是形式的“自由”氢原子,很容易通过光催化膦介导的水活化产生。巯基用作π体系自由基加氢的催化共还原剂,将最初生成的PR3-OH2自由基阳离子中间体(一个有效的质子给体)转化为自由基氢原子给体。这种共催化方法确保了水的两个H原子都可以用作还原π体系中的H原子供体。考虑到烯烃的加氢反应,引入的不含金属的PR3-OH中间体具有与已建立的以过渡金属为基础的体系相当的反应活性,这些体系参与MHAT反应。分子内或分子间氢原子转移到以碳原子或杂原子为中心的反应性原子。
参考文献
Jingjing Zhang, et al. (2023): Photocatalytic phosphine-mediated water activation for radical hydrogenation. Nature. doi.org/10.1038/s41586-023-06141-1.
Source: https://www.uni-muenster.de/en/