- 热门化合物 :
- 物质11
- 2-丙炔-1-醇
- americium,trifluoride
- neptunium hexafluoride
- neptunium tetrachloride
双原子催化剂析氧反应机理提高反应活性!
原子级精确的团簇催化剂弥合了均相催化和异相催化之间的差距,有助于全面阐明催化过程中的结构-活性关系。然而,金属纳米团簇中非单个活性位点之间的协同效应很少被探索。
原子级精确的团簇催化剂弥合了均相催化和异相催化之间的差距,有助于全面阐明催化过程中的结构-活性关系。然而,金属纳米团簇中非单个活性位点之间的协同效应很少被探索。
鉴于此,近期由中国科学院青岛生物能源与过程技术研究所(QIBEBT)的孙晓燕教授领导的研究团队在《自然通讯》上发表了题为“Synergy of dual-atom catalysts deviated from the scaling relationship for oxygen evolution reaction”的研究成果。
研究概述
该研究采用大尺度密度泛函理论探讨*O-*O耦合机制的可行性,通过提高n掺杂石墨烯负载的Fe-、Co-、Ni-和cu -异核双原子催化剂M ' M@NC的催化性能,规避结垢关系。根据所构建的活性图,可以得到合理设计的描述符来预测同核催化剂。七种异核和四种同核双原子催化剂具有高活性,超过了最小理论过电位。揭示了有利于*O-*O偶联机制从而提高反应活性的化学和结构起源。这项工作不仅对反应机理的基本认识提供了额外的见解,而且为加速发现高效催化剂提供了指导。图文导读
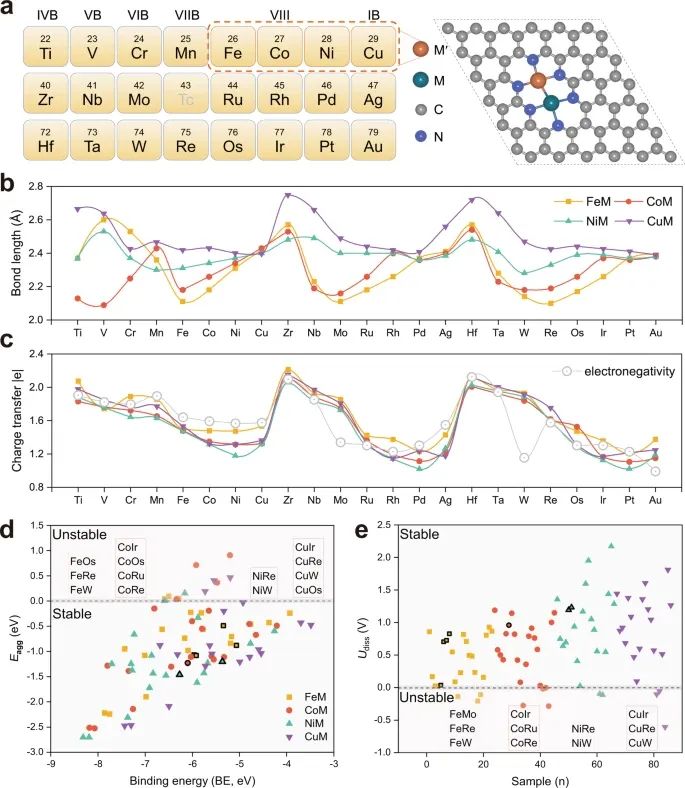
研究考虑到FeM, CoM, NiM和CuM的合成策略已经在最近的实验研究中得到了广泛的报道,本文选择Fe, Co, Ni和Cu作为M '的候选材料。与此同时,M的金属掺杂从IVB到IB的3d、4d、a和5d过渡金属(Tc除外),共82个异核dac。如图1a所示,dac中的两个过渡金属原子(M2)结合并被三个吡啶sp2-N原子包围。
先前的理论研究表明,这是在n掺杂石墨烯中嵌入M2二聚体的最有利的能量配置,各种先进的表征技术全面证实了原子分散的M2在n掺杂石墨烯上以及原子对的存在31。在此基础上,合理构建了M ' M@NC催化剂模型,其金属-金属键长在2.09 Å (CoV) ~ 2.75 Å (CuZr)之间(图1b)。部分M2二聚体的键长排列为CuM > NiM > CoM > FeM,与Fe、Co、Ni和Cu的原子半径有关,而M2二聚体的其余键长与原子半径无关。此外,M2二聚体之间的键长计算与最近的实验测量结果相吻合,例如,NiZn@NC中Ni-Zn对的键长由实验和DFT计算得到的键长为2.5 Å31, Ni-Cu键长实验观测值为2.40 Å,这也与研究的计算值2.409 Å63一致。从而验证了理论方法的合理性和DFT方法的可靠性。显然,具有适当键长的双金属活性位点有利于促进o - o - o - c - o的低能垒耦合。
图1 :M ' M@NC的结构原型、键长、电荷转移和稳定性
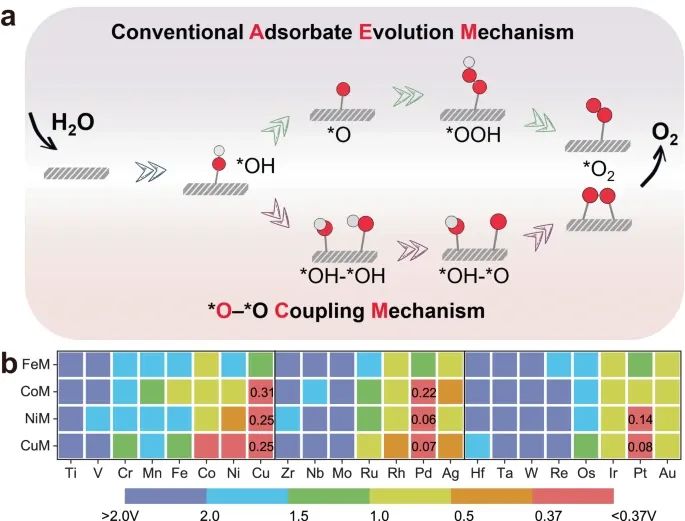
在单一活性中心进行的传统AEM中,OER涉及多个氧反应中间体,包括*OH, *O和*OOH,如图2a所示。这些中间体的结合能高度相关,导致最小理论过电位约为0.37 V60。相反,引入第二个活性中心的OCM可以打破对关键氧化中间体的结构限制。在这种情况下,最显著的区别(图2a)是,相邻的两个氧原子被吸附后,通过步骤方程得到了两个氧原子之间的相互作用力,即通过两个氧原子之间的相互作用力,得到了两个氧原子之间的相互作用力,得到了两个氧原子之间的相互作用力。2 *OH吸附的顺序,以及M2二聚体上的去质子化,遵循能量有利的构型,并最终释放耦合氧分子进入下一个反应循环(补充表14-29)。原则上,在没有*OOH中间体参与的情况下,OCM可以绕过标度关系,凭借双金属活性位点的优势成为非常有利的反应途径。
为了建立OER活性和关键中间体吸附等一系列关键因素的数据库,基于预选的OCM途径,系统地探索了86种DACs体系(FeM, CoM, NiM和CuM)的势能面。以AEM机制的瓶颈(η = 0.37 V)为基准,以热图的形式显示了高性能OER催化剂的活性趋势,如图2b所示。在η < 0.37 V条件下筛选出8种符合该基准的dac,结果表明:NiPd (0.06 V) < CuPd (0.07 V) < CuPt (0.08 V) < NiPt (0.14 V) < CoPd (0.22 V) < NiCu (0.25 V) = CuCu (0.25 V) < CoCu (0.31 V),对OER具有明显的催化活性,计算出的氧化中间体总能量、零点能量修正和熵贡献能见。有趣的是,在热图中也可以清楚地看到,从左到右,红色/蓝色区域的面积逐渐增加/减少,而红色区域表示较低的η。
因此,由早期过渡金属组成的M2二聚体会阻碍OER的进展,FeM@NC、CoM@NC、NiM@NC和CuM@NC都不利于性能的提高。相比之下,M2二聚体与后过渡金属,特别是这些贵金属Pd, Pt和Au,表现出优异的OER活性,并通过OCM远低于0.37 V。请注意,CoNi@NC在实验中报告了在10 mA cm - 2下具有252 mV的低过电位,并且在我们筛选的系统中包含了一个n和n,这可能与实际遵循的不同反应机制有关。
图2 :反应机理及活性图
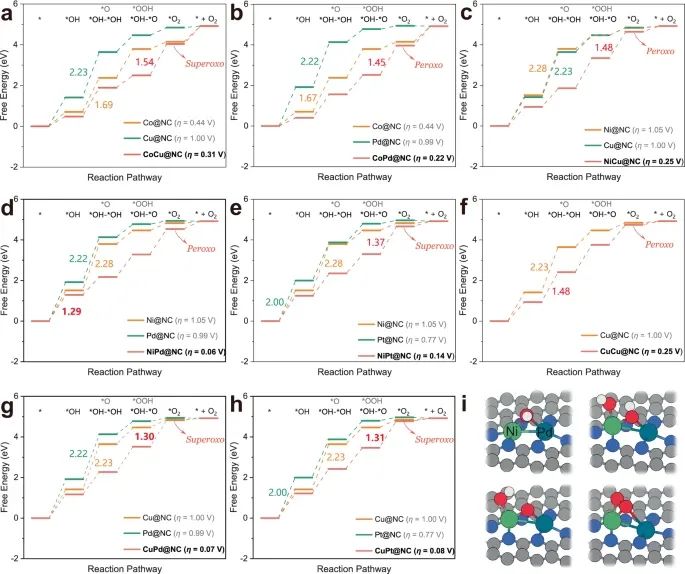
进一步将η < 0.37 V的8种M2二聚体组合与相似N配位环境下对应的M@NC (M = Co, Ni, Cu, Pd和Pt)进行比较,阐明dac和sac之间的活性差异,以及双金属活性位点的协同效应与结构工程之间的相关性(图3)。和M@NC的熵贡献能见补充表39-43。根据吉布斯自由能图的分析,可以发现沿OCM途径的dac的电位决定步骤(pds)主要发生在*O - *O耦合产生*O2(方程)。(14) o或(19)),而6个M@NC的PDS发生在*OH的去质子化生成* o(方程)。(2) o或(7))和*OH的组合(方程。(1)或(6)。值得注意的是,所有8个dac的OER活动都远高于相应的sac。以性能最好的NiPd@NC为例(图3d), 2 *OH在NiPd@NC上的吸附能量最有利。
图3 :吉布斯自由能图和优化构型
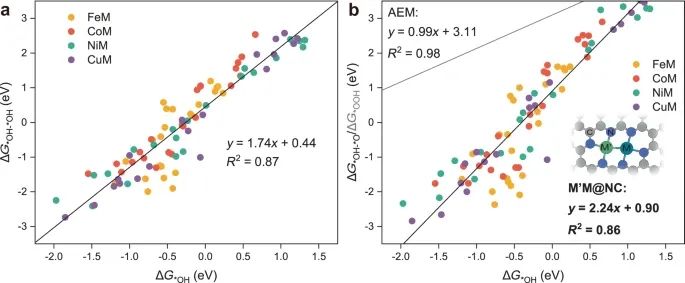
众所周知,在传统的AEM中,理想的OER催化剂的*OOH和*OH (ΔG*OOH−ΔG*OH)的无吸附能差为2.46 eV,使得所有四个基本步骤的反应自由能都在相同的量级。然而,在单个活性位点上几乎恒定的差异(ΔG*OOH−ΔG*OH)为3.2±0.2 eV,确定了最低的理论过电位为0.37 V([3.2−2.46 eV]/2)60。基于上述沿OCM的大规模筛选,8个高活性dac克服了最小理论过电位0.37 V的限制。显然,OCM的标度关系应与常规AEM的标度关系区分开来。因此,我们绘制了*OH, *OH - *OH, *OH - *O和*O2的无吸附能之间的相关性。令人感兴趣的是,*OH (ΔG*OH)的无吸附能与其他含氧中间体之间存在显著的线性相关关系(图4)。同时,双金属活性位点与图4b中具有显著挑战性的AEM的标度关系明显偏离,没有3.2±0.2 eV的恒定差值,说明双金属活性位点显著降低了0.37 V的最小理论过电位,特别是NiPd (0.06 V)、CuPd (0.07 V)和CuPt (0.08 V)。尽管M ' M@Gr上的OER活性由于氧合中间体的强吸附而较差,但临界中间体在图4b中遵循相同的标度关系,尽管具有不同的支撑,这表明OCM在dac上的一般规律。
广为接受的Sabatier原理强调吸附的中间体与催化剂活性位点的结合不能太强也不能太弱,否则会阻碍催化活性。ΔG*O−ΔG*OH通常被用作确定AEM火山地块OER活动的描述符。因此,我们采用类似于ΔG*OH - *OH的OCM描述符来描述OER在M2@NC上的催化行为。描述符的选择是基于PDS主要发生在*O - *O偶联过程中,两个*OH的吸附决定了随后的去质子化过程以及*O - *O偶联过程。有趣的是,我们发现ΔG*OH - *OH和η之间存在火山状关系(图5a),其中最佳过电位为1.56 eV <ΔG*OH - *OH < 2.46 eV,由8个η < 0的dac推导而来。V (NiP d, CuPd, CuPt, NiPt, CoPd, NiCu, CuCu和CoCu)。
图4 :OCM后的尺度关系
研究团队通过合理设计的活性描述符,进一步在预测高性能同核双原子催化剂方面取得了效率和准确性的成功。该团队还说明了OCM发生率的条件以及恒定电位法的OER性能。这些发现不仅为团簇催化剂的非单一活性位点提供了基础性见解,以提高反应性能,而且有助于为除OER之外的其他电化学反应催化剂的合理设计提供理论指导。这次应用高通量DFT进行非常规OCM研究提供了前所未有的洞察和理解。此外,更为复杂的计算方法使预测更加贴近实际的电化学条件。
参考文献
Cong Fang et al, Synergy of dual-atom catalysts deviated from the scaling relationship for oxygen evolution reaction, Nature Communications (2023). DOI: 10.1038/s41467-023-40177-1