- 热门化合物 :
- 物质11
- 2-丙炔-1-醇
- americium,trifluoride
- neptunium hexafluoride
- neptunium tetrachloride
配体控制的NiH催化烯酰酰胺的区域分化和对映选择性氢胺化
1,n-二胺(n = 1,2,3),特别是手性的二胺,是医药、农药、天然产物等领域广泛应用的重要组成部分。此外,对映体富集的1,2-或1.3-二胺长期以来也...
1,n-二胺(n = 1,2,3),特别是手性的二胺,是医药、农药、天然产物等领域广泛应用的重要组成部分。此外,对映体富集的1,2-或1.3-二胺长期以来也被认为是立体选择性过渡金属和有机催化中的特权手性配体。鉴于此,人们对其立体选择性制备作出了巨大的努力。例如,虽然已经建立了典型的催化不对称方法(如添加亚胺,烯烃二胺,开环氮吡啶等)来快速获取手性1,2-二胺,但仍然需要开发更新颖和高效的方法。
通过金属链行走的远端烯烃氢功能化最近成为一个可靠的方法,用于直接和位点选择性地安装在未激活的C(sp3)−H位点而不是初始的烯烃位点。假设烯基胺或其替代物的催化氢胺化将构成一种新的和吸引人的策略,通过链行走或非链行走过程获得1,n-二胺,特别是从相同和容易获得的烯烃。然而,由于不同烷基−金属中间体的相似反应活性难以区分,因此实现这种方法是非常困难的。Hong课题组最近实现了NiH催化的具有双齿吡啶酰胺导向基团的烯基胺的迁移δ-胺化,这被认为有助于形成热力学上更好的六元金属环物种I,以提供1,4-二胺衍生物(方案1a)。然而,N,N-双齿PA基团的强配位使其成为发展位点选择性远端氢胺化及其不对称反应的一个具有挑战性的问题。因此,作者设想合适的单齿导向基团和不同的锚定配体的协同使用将调节金属基中间体在链行走过程中特定位置的稳定性,从而构成可预测的和区域发散的烯烃氢胺化的可行策略(方案1b)。此外,手性配体的使用可能会带来不对称诱导的可能,这是目前催化不对称迁移氢功能化的一个具有挑战性的任务。手性配体必须同时促进链行走和之后的对映选择性偶联。为了控制金属链行走过程中的对映体选择性,对映体决定步骤可能是外消旋烷基−金属物种的选择性偶联生成单一的对映体.
在此,作者展示了一种弱定向基团辅助、配体控制的策略,以实现烯烃胺在初始烯烃或远程位点的区域分化氢胺化(方案1c)。令人兴奋的是,在NiH催化下,各种天然酰胺可以作为引导基团,选择性地提供有价值的α-、β-或γ-区域异构体。此外,建立了对映选择性合成手性1、2-二胺和1、3-二胺的有效方法。最近,Hong等和Shu等报道了烯丙基胺和同烯丙基胺的对映选择性氢胺化,但没有迁移。迁移的α-或β选择性氢胺化和对映选择性迁移的β选择性氢胺化尚未见报道。作者的方案还为1,1-二胺的合成提供了一个补充平台。
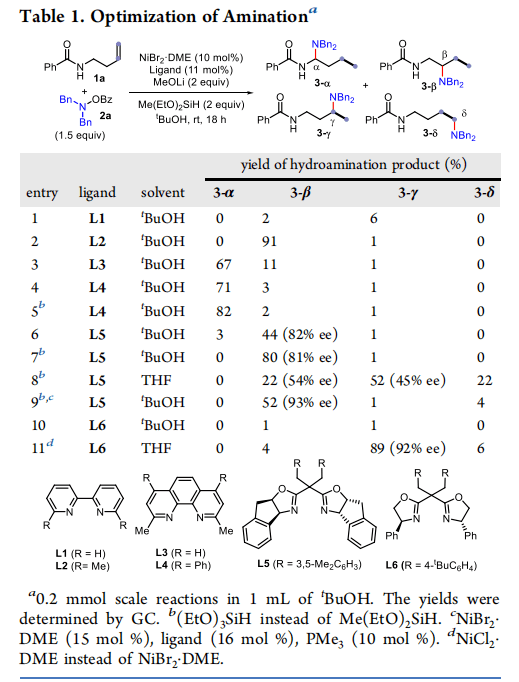
选择高烯丙基酰胺1a和邻苯甲酰羟胺(N−O)亲电试剂2a为模型底物,确定了区域收敛加氢胺化的最佳条件。事实上,在NiH催化的条件优化过程中,实现理想的(迁移)氢胺化的优良区域选择性和对映选择性非常重要,因为可以检测到四种区域异构体。经过对各种反应参数的广泛筛选,金属源、配体和溶剂对这种转化的效率和区域选择性起着至关重要的作用。如表1所示,条目1−4,使用廉价6,6’-二甲基-2,2’-联吡啶L2或2,4,7,9-四甲基-1,10-菲罗啉L4配体催化主要提供所需的β-或α-胺产品3-β或3-α,分别具有高选择性和产量。从Me(EtO)2SiH到(EtO)3SiH的氢硅烷的细微调整进一步提高了效率(条目5)。为了实现具有挑战性的对映选择性迁移氢胺化,作者发现使用双恶唑啉L5得到了一个β-选择性产物,产率为44%,ee为82%(条目6)。醚溶剂如THF,能更好促进初始烯烃部位的胺化(入口7和入口8)。最终,经过广泛的研究,添加10摩尔%的PMe3进一步提高3-β的对映选择性到93%中等产量(入口9),这是为了抑制背景非不对称反应。幸运的是,γ-胺化的选择性形成已被证明是可能的,因为在THF中使用手性二苄基取代的Ph-Box配体L6,3-γ的产率为89%,ee为92%(条目11)。
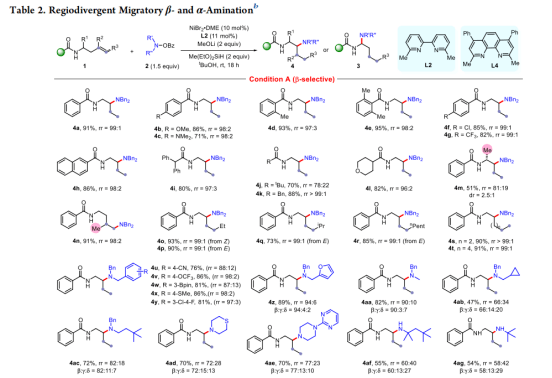
在优化的条件下,首先评估了β-位置迁移胺化的范围。如表2所示,供电子和吸电子的芳基芳香酰胺对产量和区域选择性几乎没有影响,和产物(4a−4h)可以容易分离与71−95%收益率和99:1 rr。具有不同取代基的脂肪族酰胺(4i−4l)在标准条件下也能相容。在α位的含烯烃甲基取代成功地得到了具有抗非对映选择性(2.5:1)的β选择性产物(4m),这源于近端空间位阻。主要非对映体的相对立体化学是通过对类似产物4m‘的x射线晶体学分析建立的。作者还测试了β-苯基和丁基取代的末端烯烃;然而它们没有得到任何想要的产物,而是进行了氢化反应。特别有趣的是,1,1-二取代烯烃反应得到了反马尔科夫尼科夫产物(4n),这可能是由于对NiH插入的空间效应。此外,内部烯烃(4o−4r)也适用于该反应,且双键的几何形状对反应活性没有影响(4o vs 4p)。除了同烯丙基酰胺外,链的行走还可以通过延伸的碳链而不侵蚀结果(4s−4t)。在胺化源方面,具有多种官能团和糠基的二苄基取代羟胺亲电试剂具有良好的反应活性和良好的区域选择性(4u−4z)。对于仅带有一个苄基的N−O试剂,基于取代基(4aa−4ac),区域选择性显示出很大的差异。作者认为区域选择性可能是由于氧化加成的相对速率和链行走过程,而不同的胺基源对镍的氧化加成速率不同。作者的方案也与环状(4ad和4ae)和初级胺基源(4af和4ag)兼容,尽管具有较低的区域选择性。
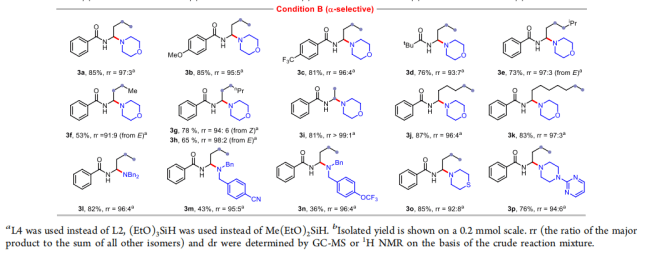
随后,作者检查了远程α-选择性氢胺化的范围(表2的底部)。含芳香族和脂肪族酰胺的底物均反应良好,得到有价值的偕-二胺产物(3a−3d)。对不同链长的内部烯烃和末端烯烃进行了测试(3e−3k),发现其反应性和区域选择性对迁移距离较长的烯烃不敏感。与具有代表性的N−O亲电试剂进行反应,得到了所需的产物(3l−3p),产率为36−85%,具有良好的区域选择性(92:8~96:4)。
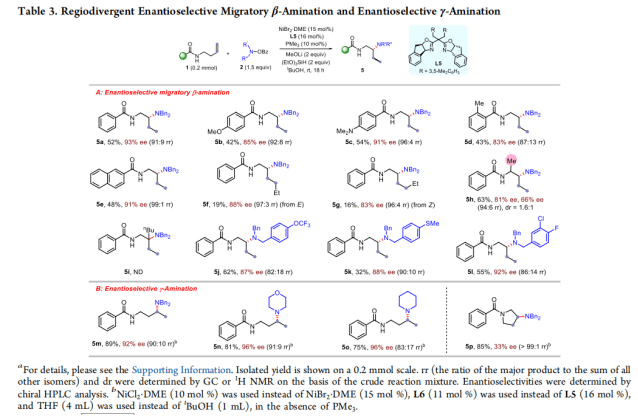
在此之后,检查了关于对映选择性迁移β-胺化的底物范围(表3A)。测试的含有甲氧基、氨基、硫甲基或三氟甲氧基的底物,它们都具有良好的区域选择性和对映选择性,产率适中(5a−5l)。添加10 mol % PMe3是有手性的。内部烯烃,无论Z/E构型如何,都可以以类似的ee传递对映转化产物,但效率较低(5f和5g)。α-取代烯烃反应顺利,得到β-选择性产物,而β-取代烯烃不适合该反应。对于对映选择性γ-胺化,采用了3种典型取代基的胺化试剂2进行测试,分离得到所需产物(5m−5p),产率为75−89%,具有良好的ee和区域选择性(表3B)。在A或B条件下环烯烃不是有效的底物,但在修正的γ-胺化条件下成功地获得了β-选择性产物,产率为85%,ee为33%。此外,目前的条件也适用于烯丙基酰胺的高度对映性和区域选择性的β-氢胺化。与Hong等和Shu等使用活性更高的O-(4-二甲基胺)苯甲酰羟胺作为亲电试剂相比,作者使用了简单的苯甲酰取代胺化试剂。
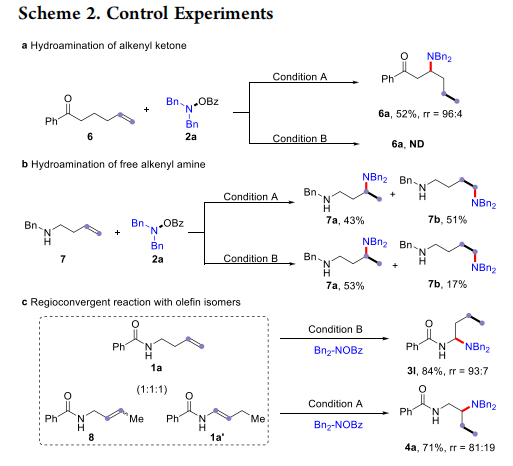
为了阐明酰胺导向基团的螯合效应,作者进行了一系列的对照实验。γ,δ-烯基酮6在A条件下的氢胺化得到了迁移的β-选择性产物,而在B条件下的反应进行了完全的加氢反应(方案2a)。非碳基N-苄基同烯丙胺7在A和B条件下,得到马式产物7a和反马式产物7b的混合物(方案2b)。联合实验表明,碳基和氮原子在区域发散氢胺化过程中都是不可或缺的。令我们高兴的是,当在B条件下使用丁替那明异构体的混合物时,获得了高产率和rr的α选择性产物(方案2c)。同时,可以获得β选择性产物,产率高,但rr较低。
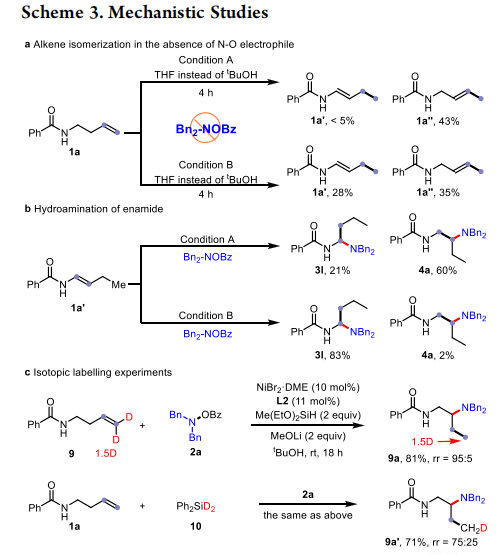
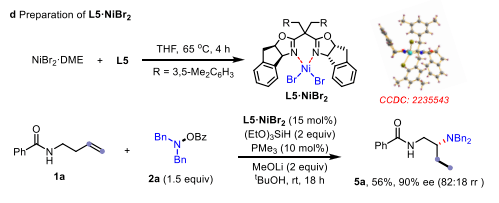
为了进一步阐明其机理,作者在烯烃1a中进行了不添加N−O亲电试剂的烯烃异构化测试(方案3a)。烯丙酰胺1a″主要在条件A下获得,而条件B产生了烯酰胺1a‘和烯丙酰胺1a″的混合物。假设配体控制的区域选择性可能归因于链行走过程的速率识别。为了识别反应中间体,烯酰胺1a‘采用条件A和条件B(方案3b)。化合物3l在B条件下顺利获得,而条件A提供了4a(产率60%)和3l(产率21%)的混合物。因此,作者得出结论,β-选择性反应可能是通过烯丙酰胺的马尔科夫尼科夫氢胺化而不是烯酰胺的反马尔科夫尼科夫氢胺化而发生的。此外,氘化底物9被用于迁移的β-胺化。然而,在其他新的位置上没有观察到氘的扰动(方案3c)。此外,氘化硅烷10的使用只在末端的甲基部分导致氘化。这些结果表明,NiH的迁移可能是不可逆的。然而,通过将L5与NiBr2·DME的简单混合,得到了L5·NiBr2的晶体结构(方案3d)。此外,使用这种晶体用于模型β-胺化也可以提供产品5a,其结果与最佳条件相似。
综上所述,通过配体控制和定向基团辅助策略实现了镍催化的三位选择性的烯基胺氢胺化。可预测的位点选择方案与不同的天然酰胺导向基团兼容,它们提供一系列结构多样的1,1-,1,2-和1,3-二胺,具有良好到优秀的区域选择性。此外,还公开了对映选择性迁移β-氢胺化,从而能够传递具有高价值的手性1,2-二胺。通过迁移或非迁移反应形成连接的五、六或七元镍酰环被认为是控制区域选择性的关键步骤。
https://pubs.acs.org/10.1021/acscatal.3c01845